
Consensus Guidelines for Juvenile Dermatomyositis Management Save

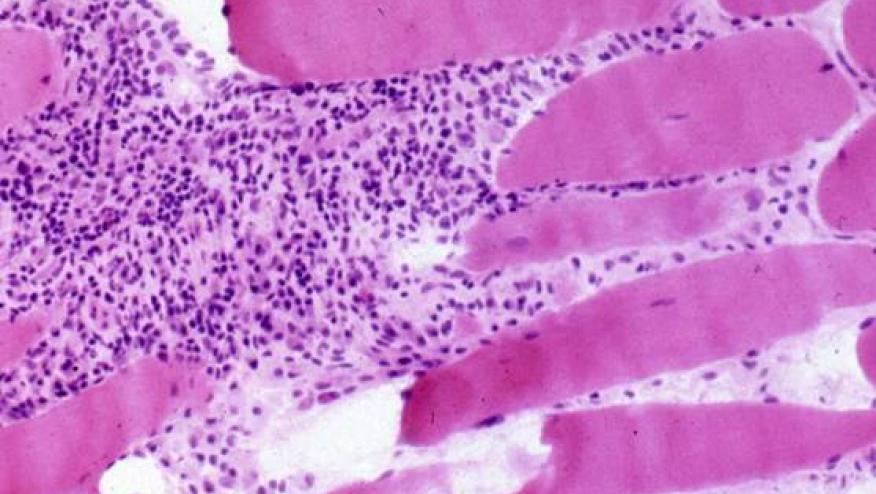
SHARE (Single Hub and Access point for pediatric Rheumatology in Europe) was established in 2012 to optimise the diagnosis and treatment of several pediatric diseases. They have recently published their recommendations regarding the diagnosis and management of juvenile dermatomyositis (JDM).
Recommendations were developed by an evidence-informed consensus process involving 19 experienced worldwide pediatric rheumatologists and 2 experts in pediatric exercise physiology and physical therapy, mainly from Europe. Recommendations were accepted if >80% consensus was reached.
In summary, there are 7 overarching principles, 33 recommendations on diagnosis and 19 recommendations on therapy. Here are the highlights from their article.
Overarching Principles:
- All children with suspected idiopathic inflammatory myopathies should be referred to a specialised centre for JDM
- High-risk patients need immediate/urgent referral to a specialised centre. High risk patients includes the following: severe disability, presence of aspiration or dysphagia, GI vasculitis, myocarditis, parenchymal lung disease, CNS disease, skin ulceration, or age <1 year
- For JDM, validated outcome tools should be used to measure health status (Childhood Health Assessment Questionnaire, patient/parent visual analogue scale, Childhood Health Questionnaire, Juvenile Dermatomyositis Multi-dimensional Assessment Report)
- All children with JDM should have disease damage assessed at least yearly using a standardised disease damage measure, such as the Myositis Damage Index
- All JDM patients should have the opportunity to be registered within a research registry/repository.
DIagnostics Recommendations:
- Alternative diagnoses should be considered including metabolic or mitochondrial myopathies and dystrophies.
- In every JDM patient, consider muscle enzymes—CPK, LDH, AST, ALT, adolase, CBC, ESR, CRP, Myositis-specific and myositis-associated antibodies, TFTs, Vitamin D, UA, nailfold capillaroscopy, ECHO, ECG, PFTs, CXR or HRCT, MRI of muscules or quantitative ultrasound, EMG, Muscle biopsy
- MRI can an aid to diagnosis and should be interpreted by an expert radiologist
- Muscle biopsy should be done in all cases where the presentation of JDM is atypical; or in the absence of rash/skin signs. (there is insufficient evidence to recommend a needle biopsy as opposed to an open biopsy in children.
- If MRI or muscle biopsy is not possible, increased muscle echo intensity on muscle ultrasonography may be indicative of myositis
- Swallow function should be formally assessed in every patient.
- EMG or nerve conduction velocity should be considered to differentiate myopathy from neuropathy when diagnosis of JDM is uncertain. E
- MG does not detect metabolic myopathies reliably and further workup is required if this diagnosis is suspected.
- At time of diagnosis or disease flare, standardised nailfold capillaroscopy assessment is recommended.
- All patients with JDM should have an assessment of lung involvement at time of diagnosis, including PFTs, with CO diffusion. If pulmonary function tests are indicative of interstitial lung disease, further investigations (CXR/ HRCT) are needed.
- All patients with JDM should have echocardiography and ECG at diagnosis.
- Calcinosis should be looked for in all patients with JDM.
- We recommend use of muscle enzymes (CPK, LDH, AST) for diagnosis and disease monitoring in JDM, although it must be recognised muscle enzymes may be normal despite active disease.
- There is no significant diagnostic benefit gained from measurement of antinuclear antibody in JDM.
- But, measurement of myositis-specific autoantibodies (such as anti-TIF 1-γ (p155), anti-NXP2/(p140/MJ), anti-MDA5 and anti-SRP) should be considered, when available.
- In patients with overlap features, measurement of myositis-associated-antibodies such as anti-PmScl, anti-U1-RNP, anti-La (‘SSB’), anti-Ro (‘SSA’) and anti-Sm may be helpful to clarify the diagnosis.
Treatment Recommendations:
- Sun protection, including the routine use of sunblock should be encouraged for patients with JDM.
- Involve a multidisciplinary team when treating patients with JDM (physiotherapist, nurse specialist, etc)
- We recommend the induction regimen for treatment of new onset patients with JDM to be based on high dose of corticosteroids (oral or intravenous) combined with MTX.
- High-dose corticosteroids should be administered systemically either orally or intravenously in moderate–severe JDM.
- High-dose corticosteroids should be administered intravenously if there are concerns about absorption.
- Addition of MTX or ciclosporin A leads to better disease control than prednisolone alone; safety profiles favour the combination of methotrexate and prednisolone.
- MTX should be started at a dose of 15–20 mg/m2/week (max absolute dose of 40 mg /week) preferably administered subcutaneously at disease onset
- If a newly diagnosed patient has inadequate response to treatment, intensification of treatment should be considered within the first 12 weeks- Intravenous immunoglobulin may be a useful adjunct for resistant disease, particularly when skin features are prominent
- Mycophenolate (MMF) may be a useful therapy for muscle and skin disease (including calcinosis).
- Ongoing skin disease reflects ongoing systemic disease and therefore should be treated by increasing systemic immunosuppression. Topical tacrolimus (0.1%)/topical steroids may help localised skin disease, particularly for symptomatic redness or itching.
- Methotrexate alternatives include ciclosporin A or MMF.
- For patients with severe disease (such as major organ involvement/extensive ulcerative skin disease), addition of intravenous cyclophosphamide should be considered.
- B cell depletion therapy (rituximab) can be considered as an adjunctive therapy for those with refractory disease. Clinicians should be aware that rituximab can take up to 26 weeks to work.
- Anti-TNF therapies can be considered in refractory disease; infliximab or adalimumab are favoured over etanercept.
- In the presence of developing or established calcinosis, intensification of immunosuppressive therapy should be considered.
- There is no high-level evidence of when to stop therapy; however, consideration may be given to withdrawing treatment if a patient has been off steroids and in remission on methotrexate (or alternative DMARD) for a minimum of 1 year.
ADD THE FIRST COMMENT
Disclosures
The author has no conflicts of interest to disclose related to this subject

If you are a health practitioner, you may Login/Register to comment.
Due to the nature of these comment forums, only health practitioners are allowed to comment at this time.