
Consensus Recommendations for Juvenile Dermatomyositis Save

SHARE (Single Hub and Access point for pediatric Rheumatology in Europe) is a group established in 2012 to develop and optimize diagnostic and management regimens in Europe for children and young adults with rheumatic diseases.
As a part of this activity, a committee of 19 pediatric rheumatologists and others (exercise physiology and physical therapy) were convened to formulate recommendations for management of juvenile dermatomyositis (JDM) using the European League Against Rheumatism (EULAR) standard methods for developing best practice.
After a validated systematic literature review, and an online survey, there were two consensus meetings that used nominal group technique to develop recommendations requiring >80% agreement.
The SHARE initiative identified 7 overarching principles, 33 recommendations on diagnosis and 19 recommendations on therapy on topics that assessment of skin, muscle and major organ involvement and suggested treatment pathways for JDM. These incude the following:
Overarching Principles
- All children with suspected idiopathic inflammatory myopathies should be referred to a specialised centre
- High-risk patients need immediate/urgent referral to a specialised centre.
- Validated tools should be used to measure health status
- All children with JDM should have disease activity (muscle, skin, major organ) assessed regularly in a standardised way, using tools such as the Disease Activity Score
- All patients with JDM should have the opportunity to be registered within a research registry/repository
Recommendations Regarding Diagnosis
- In every patient in whom a diagnosis of JDM is considered, the following investigations should be considered:
- Muscle enzymes, ESR, CRP, myositis-specific and myositis-associated antibodies, infection screen, consideration of tests for metabolic/mitochondrial myopathies (especially in the absence of rash/atypical presentation), urine dipstick, nailfold capillaroscopy, echocardiogram and ECG, pulmonary function tests (chest X-ray and HRCT if concern), MRI of muscles (+quantitative ultrasound), EMG (particularly if suspicion of neuropathy/disorder of neuromuscular junction), muscle biopsy (especially in the absence of rash/atypical presentation) - Both muscle strength and function should be tested at diagnosis and follow up by formal validated measures
- MRI can be used to aid diagnosis of JDM.
- MRI can be used to help monitor disease activity.
- When used, MRI should be carried out by defined protocols that enhance detection of muscle inflammation, such as T2 weighted/STIR sequences.
- MRI should be interpreted by an expert radiologist.
- A muscle biopsy should be done in all cases where the presentation of JDM is atypical; in particular in the absence of rash/skin signs.
- If a muscle biopsy is performed for diagnosis of JDM, a standardised JDM biopsy score tool should be used to quantify severity of histological abnormalities.
- Expert histopathological opinion is required to define features of inflammation in JDM muscle biopsy.
- When doing a muscle biopsy, there is insufficient evidence to recommend a needle biopsy as opposed to an open biopsy in children.
- In cases where MRI or muscle biopsy is not possible, increased muscle echo intensity on muscle ultrasonography (when performed by an experienced sonographer) may be indicative of myositis.
- Swallow function should be formally assessed in every patient. The assessment may include a speech and language therapy assessment, video fluoroscopy/barium studies.
- EMG or nerve conduction velocity should be considered to differentiate myopathy from neuropathy when diagnosis of JDM is uncertain.
- EMG does not detect metabolic myopathies reliably and further workup is required if this diagnosis is suspected.
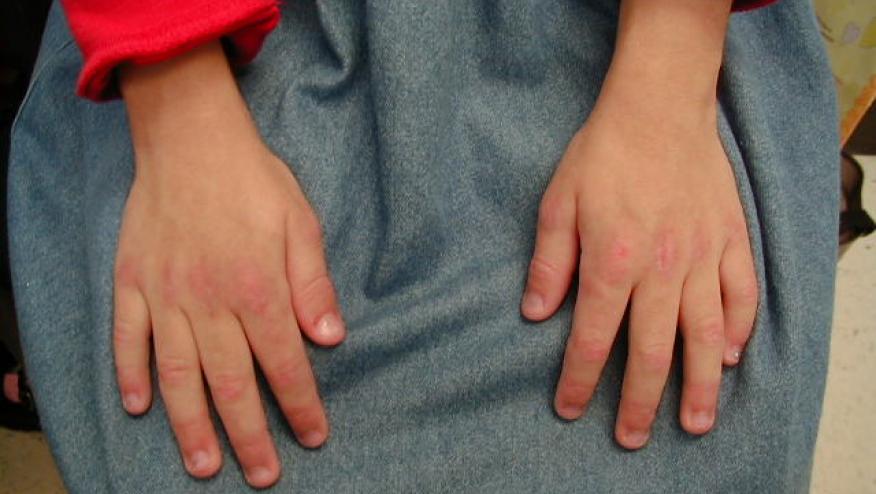
- Assessment of nailfold capillaries should be used to aid diagnosis of JDM.
- At time of diagnosis or disease flare, standardised nailfold capillaroscopy assessment is recommended. During follow-up, assessment of nailfold capillaries should be performed regularly.
- A formal CAT should be used to aid diagnosis of JDM.
- A formal CAT should be used to monitor skin disease activity over time.
- Skin tools may include the DAS (skin), MITAX (skin) or CAT.
- All patients with JDM should have an assessment of lung involvement at time of diagnosis.
- Assessment should include pulmonary function tests, including CO diffusion. If pulmonary function tests are indicative of interstitial lung disease, further investigations (CXR/ HRCT) are needed.
- All patients with JDM should have echocardiography and ECG at diagnosis.
- Patients at particular risk of cardiac dysfunction should have repeated cardiac evaluation. Risk factors include hypertension, high disease activity 1 year post diagnosis, long-term high corticosteroid burden or chronic ongoing active disease.
- Calcinosis should be looked for in all patients with JDM.
- Plain radiographs may be used for the evaluation of calcinosis.
- We recommend use of muscle enzymes (CPK, LDH, AST) for diagnosis and disease monitoring in JDM, although it must be recognised muscle enzymes may be normal despite active disease.
- Measurement of von Willebrand factor does not provide any additional information for diagnosis of JDM.
- There is no significant diagnostic benefit gained from measurement of antinuclear antibody in JDM.
- Measurement of myositis-specific autoantibodies should be considered, when available.
- In patients with overlap features, measurement of myositis-associated-antibodies such as anti-PmScl, anti-U1-RNP, anti-La (‘SSB’), anti-Ro (‘SSA’) and anti-Sm may be helpful to clarify the diagnosis.
- urther validation studies are recommended to define the use of more sensitive biomarkers in JDM.
Recommendations Regarding Treatment
- Sun protection, including the routine use of sunblock on sun-exposed areas should be encouraged for patients with JDM.
- When treating patients with JDM, it is particularly important to have a physiotherapist and a specialist nurse actively involved as part of a multidisciplinary team.
- Treatment of JDM should include a safe and appropriate exercise programme, monitored by a physiotherapist.
- We recommend the induction regimen for treatment of new onset patients with JDM to be based on high dose of corticosteroids (oral or intravenous) combined with MTX.
- High-dose corticosteroids should be administered systemically either orally or intravenously in moderate–severe JDM.
- High-dose corticosteroids should be administered intravenously if there are concerns about absorption.
- Corticosteroid dose should be weaned as the patient shows clinical improvement.
- Addition of MTX or ciclosporin A leads to better disease control than prednisolone alone; safety profiles favour the combination of methotrexate and prednisolone.
- MTX should be started at a dose of 15–20 mg/m2/week (max absolute dose of 40 mg /week) preferably administered subcutaneously at disease onset. If a newly diagnosed patient has inadequate response to treatment, intensification of treatment should be considered within the first 12 weeks, after consultation with an expert centre.
- Intravenous immunoglobulin may be a useful adjunct for resistant disease, particularly when skin features are prominent.
- MMF may be a useful therapy for muscle and skin disease (including calcinosis).
- Ongoing skin disease reflects ongoing systemic disease and therefore should be treated by increasing systemic immunosuppression. Topical tacrolimus (0.1%)/topical steroids may help localised skin disease, particularly for symptomatic redness or itching.
- In patients who are intolerant to methotrexate, change to another DMARD, including ciclosporin A or MMF.
- For patients with severe disease (such as major organ involvement/extensive ulcerative skin disease), addition of intravenous cyclophosphamide should be considered.
- B cell depletion therapy (rituximab) can be considered as an adjunctive therapy for those with refractory disease. Clinicians should be aware that rituximab can take up to 26 weeks to work.
- Anti-TNF therapies can be considered in refractory disease; infliximab or adalimumab are favoured over etanercept. 3 D 92
In the presence of developing or established calcinosis, intensification of immunosuppressive therapy should be considered. - There is no high-level evidence of when to stop therapy; however, consideration may be given to withdrawing treatment if a patient has been off steroids and in remission on methotrexate (or alternative DMARD) for a minimum of 1 year.
SHARE recommendations are based on expert opinion informed by the best available evidence and attempts to defined best practices for treatment of patients suffering from JDM.
ADD THE FIRST COMMENT
Disclosures
The author has no conflicts of interest to disclose related to this subject


If you are a health practitioner, you may Login/Register to comment.
Due to the nature of these comment forums, only health practitioners are allowed to comment at this time.