

EULAR Consensus Guidelines for Juvenile Localized Scleroderma Save

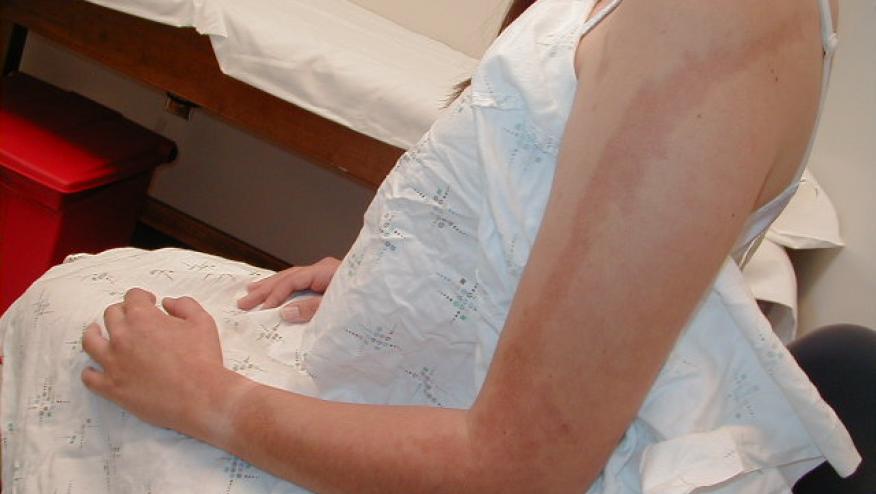
A EULAR international committee of 15 experts in pediatric rheumatology have established consensus-based recommendations for the management of juvenile localised scleroderma (JLS).
JLS is an uncommon disorder that is commonly seen by pediatric and adult rheumatologists. Evidence-based guidelines are sparse and management is mostly based on physicians’ experience.
These recommendations come from experienced pediatric rheumatologists and two young fellows, based on a validated systematic literature review, an online survey and two consensus meetings. Recommendations required ≥80% agreement.
This guidance has 1 overarching principle, 10 recommendations on assessment and 6 recommendations on therapy covering the assessment of skin and extracutaneous involvement and treatment pathways. In assessing skin lesions they advocate for the use of the LoSCAT (Localized Scleroderma Cutaneous Assessment Tool) - a scoring system that includes a Skin Severity Index (LoSSI) and a Skin Damage Index (LoSDI).
Overarching principle
All children with suspected localised scleroderma should be referred to a specialised paediatric rheumatology centre.
Recommendations regarding diagnosis and assessment
- LoSSI, which is part of LoSCAT, is a good clinical instrument to assess activity and severity in JLS lesions and is highly recommended in clinical practice.
- LoSDI, which is part of LoSCAT, is a good clinical instrument to assess damage in JLS and is highly recommended in clinical practice.Infrared thermography can be used to assess activity of the lesions in JLS, but skin atrophy can give false-positive results.
- A specialised US imaging, using standardised assessment and colour Doppler, may be a useful tool for assessing disease activity, extent of JLS and response to treatment.
- All patients with JLS at diagnosis and during follow-up should be carefully evaluated with a complete joint examination, including the temporomandibular joint.
- MRI can be considered a useful tool to assess musculoskeletal involvement in JLS, especially when the lesion crosses the joint.
- It is highly recommended that all patients with JLS involving face and head, with or without signs of neurological involvement, have an MRI of the head at the time of the diagnosis.
- All patients with JLS involving face and head should undergo an orthodontic and maxillofacial evaluation at diagnosis and during follow-up.
- Ophthalmological assessment, including screening for uveitis, is recommended at diagnosis for every patient with JLS, especially in those with skin lesions on the face and scalp.
- Ophthalmological follow-up, including screening for uveitis, should be considered for every patient with JLS, especially in those with skin lesions on the face and scalp.
Recommendations regarding treatment
- Systemic corticosteroids may be useful in the active inflammatory phase of JLS. At the same time as starting systemic corticosteroids, MTX or an alternative DMARD should be started.
- All patients with active, potentially disfiguring or disabling forms of JLS should be treated with oral or subcutaneous methotrexate at 15 mg/m²/week.
- If acceptable clinical improvement is achieved, methotrexate should be maintained for at least 12 months before tapering.
- Mycophenolate mofetil may be used to treat severe JLS or MTX-refractory or MTX-intolerant patients.
- Medium-dose UVA1 phototherapy may be used to improve skin softness in isolated (circumscribed) morphoea lesions.
- Topical imiquimod may be used to decrease skin thickening of circumscribed morphoea.
Abbreviations: JLS, juvenile localised scleroderma; L, level of evidence; LoSCAT, Localized Scleroderma Cutaneous Assessment Tool; LoSDI, Localized Scleroderma Skin Damage Index; LoSSI, Localized Scleroderma Skin Severity Index; S, strength of recommendation; US, ultrasound.
ADD THE FIRST COMMENT
Disclosures
The author has no conflicts of interest to disclose related to this subject




If you are a health practitioner, you may Login/Register to comment.
Due to the nature of these comment forums, only health practitioners are allowed to comment at this time.