

Monocyte Patrolling Contributes to Lupus Glomerulonephritis Save

Systemic lupus erythematosus (SLE) is an autoimmune disease with a propensity to develop glomerulonephritis (47-70%) or end-stage kidney disease despite therapy.
Hallmarks of the disease are the appearance of immune complexes (IC) containing autoreactive Abs and TLR-activating nucleic acids, whose deposition in kidney glomeruli is suspected to promote tissue injury and glomerulonephritis (GN).
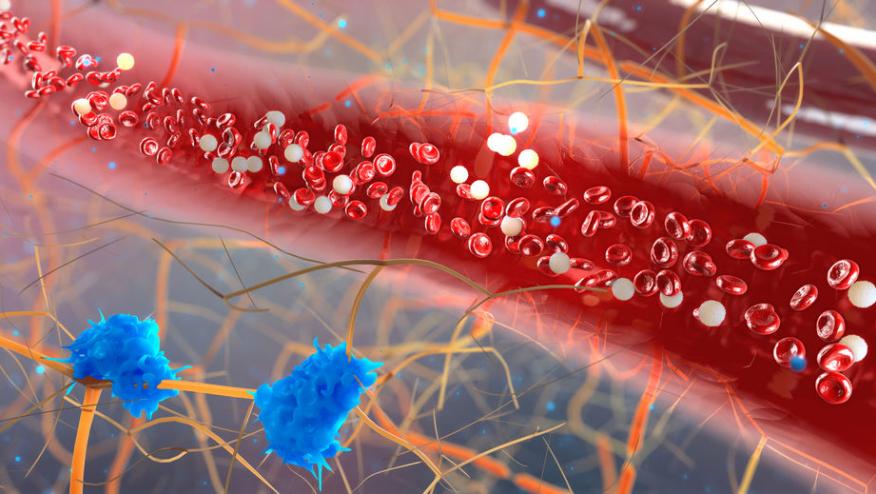
The pathogenesis of lupus GN was investigated using 3 different mouse models of SLE and SLE patients all of whom demonstrated glomerular accumulation of patrolling monocytes (PMos), a cell type with an emerging key function in vascular inflammation.
Lupus nephritis is considered an immune complex–mediated (IC-mediated) form of GN. Although the relevance of ICs in lupus nephritis is debated, IC deposition cand lead to complement-mediated cytotoxicity and end-organ damage.
ICs may also contribute to GN by activating Toll-like receptors (TLR), including TLR7, TLR8, and TLR9, on B cells, further contributing the tissue damage and inflammation. It is possible that the therapeutic efficacy of antimalarials are mediated by the prevention of TLR activation.
A potential B cell–independent mechanism for lupus GN is supported by more than 50 genetic SLE susceptibility loci with functions outside the adaptive immune system. One such susceptibility locus for SLE and lupus GN TNIP1 (TNFAIP3-interacting protein 1; also referred to as ABIN1, A20-binding inhibitor of NF-κB activation 1). ABIN1 is a negative regulator of the TNFR, TLR, and IL-17R pathways, and, ABIN1-deficient mice develop a systemic lupus-like disease that includes major characteristics of human SLE, including autoreactive Abs and severe GN.
Studies reported in the Journal of Clinical Investigation have shown that monocyte-specific deletion of ABIN1, promoted kidney disease, whereas selective elimination of patrolling monocytes provided protection against GN.
Patrolling monocytes (PMos) are one of the two monocytes found in the peripheral blood (PB), PMos numbers are increased in the PB and kidney glomeruli of lupus. Investigators have shown that PMos are capable of mediating tissue injury in lupus-nephritis based on studies done to characterize GN in ABIN1-deficient mice and 2 additional independent lupus models.
These data identify TLR-activated PMos as the principal component of an intravascular process that contributes to glomerular inflammation and kidney injury. based on the SLE susceptibility locus TNFAIP3-interacting protein 1 (TNIP1, also known as ABIN1).
ADD THE FIRST COMMENT
Disclosures
The author has no conflicts of interest to disclose related to this subject





If you are a health practitioner, you may Login/Register to comment.
Due to the nature of these comment forums, only health practitioners are allowed to comment at this time.