

Update on Myositis Save

The ACR/ARHP 2018 national meeting in Chicago presented us with opportunities to learn more about inflammatory myositis. A particularly interesting lecture was HOT topics in myositis. There were a few abstracts on MDA-5 related disease and a few on new or repurposed medications for dermatomyositis which will be covered here.
HOT topics in myositis
Dr. Ingrid Lundberg reviewed aspects of inflammatory myositis including the relatively new 2017 EULAR/ACR classification criteria for idiopathic inflammatory myositis. She reminded us that a normal CK and even normal muscle biopsy do not exclude the diagnosis of dermatomyositis if the clinical index of suspicion is high. While the MMT8 (manual muscle testing), muscle enzymes, and functional measures can help determine disease activity, if a patient’s strength and even CK is normal on motor strength testing but they have easy fatigability, performing a functional index in myositis may be helpful (the FI uses repetitive and endurance measures and is thus more sensitive to muscle disease).
Overlap myositis
Dr. Julie Paik went on to discuss overlap myositis, with systemic sclerosis being the most common connective tissue disease associated with myositis (next being SLE and RA).
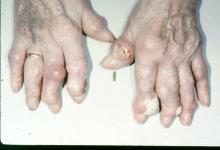
Up to 25% of patients with systemic scleroderma develop skeletal myopathy, which has been increasingly recognized as a poor prognostic feature. This occurs more commonly among African Americans, those with diffuse cutaneous and more severe disease. Muscle biopsies of patients with scleroderma myositis is most commonly notable non-specific or necrotizing myositis. Acute neurogenic atrophy, however, can be seen in almost half of patients.
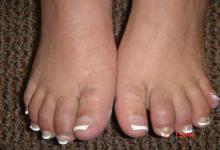
An unusual, and relatively unique, feature among patients with scleroderma myositis that has recently been described is a ‘fibrosing myopathy’ with fibrosis of the endomyseal and perimyseal tissue without inflammatory infiltrates. This pattern has been associated with African American race, short disease duration, U3-RNP antibody, anti-topoisomerase, and high mortality rates, with a high rate of death from non-ischemic cardiomyopathy (cardiac MRIs may in fact show myocardial fibrosis). Notably, patients with this pattern of myositis often have lower CK and aldolase elevation and an elevated troponin; thus, a relatively mild CK elevation should not be ignored in systemic scleroderma.
“Rheumatoid myositis” likely describes a group of patients with anti-synthetase syndrome and positive ACPAs. As we recall, about two thirds of patients with an anti-synthetase syndrome have inflammatory arthritis at onset (with almost 20% of Jo-1 positive patients presenting with arthritis initially as their main clinical phenotype).
Patients with PM-Scl+ myositis often have more Raynaud’s, mechanic’s hands, and calcinosis than those with an anti-synthetase syndrome, and frequent ILD (as I’ve heard it said, they look more like an anti-synthetase syndrome than some anti-synthetase syndrome patients). Thus, as this antibody may not be checked as part of a standard panel, it may be helpful to specifically request it if suspicion remains high. Clinically, they may have more arm abductor than hip flexor muscle weakness and intense perivascular inflammation on muscle pathology.
Inclusion Body Myositis (IBM)
Dr. Andrew Mammen then reviewed some very interesting developments in the field of IBM after an overview of its clinical phenotype. He suggested the use of the criteria published by Lloyd et al in 2014 for diagnosis, which include: 1. Finger flexion OR knee extension weakness, 2. Endomyseal inflammation, and 3. Invasion of non-necrotic muscle fibers OR presence of rimmed vacuoles on biopsy. These have a 90% sensitivity and 96% specificity for the diagnosis of IBM.
While IBM has been thought by many to be a degenerative condition (with abnormal protein aggregates in the muscle), an autoimmune component is increasingly being recognized. Recently, it has been described that patients with IBM have a clonally expanded population of T cells that persists for years and are found in multiple muscle fibers and in the circulation.
In fact, 58% of patients with IBM fulfill criteria for T-cell large granular lymphoma (LGL), and these patients have more severe IBM. While most IBM patients don’t have a T-LGL phenotype, it is thought that persistent antigenic stimulation to an as-of-yet undetermined antigen may precipitate a neoplastic-like disorder. Open label trials depleting T cells, with anti-thymocyte globulin and alemtuzumab, have suggested benefit in IBM.
Furthermore, while B20+ B cells are not generally seen in the muscle of patients with IBM, CD138+ plasma cells are abundant, and can form an ectopic lymphoid collection. The antibody against cytosolic 5’ nucleotidase 1a (NT5C1a Ab), a protein abundant in skeletal muscle, has been associated with IBM. However, NT5C1a antibody is also found in patients with dermatomyositis, SLE, and Sjogren’s, and it is thus unclear if these antibodies are pathogenic.
At present, we have no effective therapy for IBM, with potential benefit in some subsets with methotrexate. Oxandrolate (a synthetic androgen thought to build muscle) showed possible benefit, while bimagrumab (activin receptor antibody) and arimoclomol (which decreases protein aggregation), did not meet their primary endpoint. Rapamycin, an immunosuppressive and autophagy stimulator, met some secondary endpoints. Thus, it is hoped that effective therapy is on the horizon.
MDA-5 disease
There were a few abstracts at the ACR national meeting addressing the spectrum of MDA-5 disease and its treatment.
Abstract 1331 presented by Dr. Yves Allenbach et al in France suggested 3 distinct clusters of MDA5 + disease: 1. A group with rapidly-progressive ILD with mechanic’s hands, 2. A group with arthralgia/arthritis and cutaneous disease, and 3. A group with severe cutaneous vasculopathy (Raynaud’s, skin ulceration, digital necrosis, and calcinosis) with myositis which tended to be predominantly male. Mortality outcome varied by cluster (65% vs 0% vs 19% respectively).
Another abstract 838 presented at the first plenary session by Dr. Hideaki Tsuji et al suggested that in a group of Japanese patients with rapidly progressive interstitial lung disease associated with the MDA-5 antibody, aggressive combined immunosuppressive therapy with prednisone (1 mg/kg/day for 4 weeks with a taper), with IV Cytoxan (500-1000 mg/m2 every 2 weeks for 6 doses, then every 4 weeks for total 10-15 doses), with tacrolimus to goal through of 10-12 ng/mL, with or without plasmapheresis (in the case of oxygen requirement) resulted in improved survival compared to ‘step up’ therapy. As expected, this resulted in CMV re-activation in some, but overall death rate was still statistically improved compared to historical controls.
New therapies for dermatomyositis
Additionally, there were 2 new/repurposed medications put forth for the treatment of dermatomyositis.
Abstracts 2284 and 2977 presented by Dr. Victoria Werth (et al) addressed lenabasum, a cannabinoid receptor type 2 agonist which is a non-immunosuppressive therapy that activates resolution of the innate immune response. In an open label extension trial of a phase 2 study of patients with refractory, skin-predominant dermatomyositis, no serious adverse events were seen and benefit was shown in skin scores. This medication reduced both type 1 and 2 interferon (IFN beta and IFN gamma) levels in the skin. CD4+ T cells in the skin biopsies of lenabasum-treated patients also decreased, as did IL-31 protein, which is associated with pruritis.
In another small study presented by Dr. Julie Paik et al in late breaking abstract 2, all patients who had refractory dermatomyositis (to 2 or more steroid sparing agents or high dose steroids) and were prescribed tofacitinib demonstrated efficacy at 12 weeks in a proof of concept study; most were able to taper off steroids. With a small sample size, there was a trend towards a lower CXCL 9 and 10. The more notable improvements were seen especially among patients with a TIF gamma positive auto-antibody.
Thus, exciting new developments are occurring in the field of inflammatory myositis. ACRBeyond is available for those who want to hear the lectures firsthand.




If you are a health practitioner, you may Login/Register to comment.
Due to the nature of these comment forums, only health practitioners are allowed to comment at this time.