

FDA Arthritis Advisory Committee Recommends Approval of Adalimumab Biosimilar Save


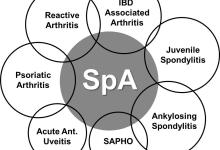
The Food and Drug Administration’s Arthritis Advisory Committee yesterday (July 12) recommended that Amgen biosimilar (ABP 501) to Humira (adalimumab) be approved for use in the United States. The panel unanimously voted, 26-0, that ABP 501 be recommended for use in the same indications as adalimumab: rheumatoid arthritis (RA), juvenile idiopathic, psoriatic arthritis, ankylosing spondylitis, plaque psoriasis, adult Crohn’s disease, and adult ulcerative colitis.
The FDA approved adalimumab in December 2002. It is currently licensed for treatment of these and other conditions. The recent approved indications for uveitis and hidradenitis suppurativa are not eligible for consideration with this new biosimilar application.
ABP 501 is the biosimilar adalimumab and targets tumor necrosis factor-alpha (TNF-α) in the management of RA and multiple inflammatory diseases. The originator product is a leader in worldwide sales, totaling $14 billion in 2015.
The Vote
The 26-member voting committee comprised healthcare professionals, including 5 rheumatologists with a wide range of scientific and analytical expertise. After extensive review of the data, voting members discussed whether sufficient biosimilarity was demonstrated between ABP-501 and adalimumab and whether there are clinically meaningful differences between the two products and if there was sufficient information given to support extrapolation to all the indications afforded the US licensed Humira product.
Background
Biosimilarity is defined by the 351(k) regulatory pathway that requires comparison of a new biosimilar to the reference product (innovator drug), wherein the biosimilar must be highly similar, with no clinically meaningful differences with safety, purity and potency.
The manufacturer presented its evidence intended to fulfill the regulatory requirement for biosimilarity between ABP-501 and adalimumab. This biologic license application included extensive based on a large body of analytical data (PK, PD, etc.), mechanism of action similarity data, and safety, immunogenicity and clinical data.
Once the biosimilar is meets standards established by the reference product, there is less need for clinical trial and safety data – since these have already been established by the reference product (e.g., Humira). Thus, one or more randomized clinical trials (with immunogenicity studies) are needed for approval. There is no defined pivotal study to establish biosimilarity. Instead of relying on numerous clinical trials, analytical similarity assumes a greater role as it forms the foundation of a biosimilar development program.
The validation of biosimilarity (focused on structure and function) dominated most of the presentations and discussion. This included similarity data on structure, folding, drug binding, peptide mapping, amino acid sequence, bioactivity, glycosylation, PK/PD studies, immunogenicity studies, etc.
Extrapolation to multiple indications based on a single indication (RA) clinical trial is a unique feature of the 351(k) regulatory pathway (also known as the abbreviated development program). Thus, the sponsor can perform a single indication clinical trial (e.g., in RA) and also request the same indications achieved by the reference product (e.g., Humira) such as Crohn’s, ulcerative colitis, psoriasis, psoriatic arthritis, ankylosing spondylitis, juvenile idiopathic arthritis, etc.). However this requires they demonstrate biosimilarity and validation of similar mechanism of action (MOA), immunogenicity and PK. Note that extrapolation does not apply to indications achieved through orphan drug approval process.
The Data
The AAC considered the data from a single-dose pharmacokinetic (PK) study and two clinical trials. The PK study (study 217) was a three-way comparison of ABP 501, US-licensed adalimumab, and European Union (EU)–approved adalimumab that supported the PK similarity of ABP 501 to the US-licensed adalimumab.
Study 262 was a 26-week, randomized, study that included 526 patients with active RA despite background methotrexate. Patients were randomly assigned to receive either ABP 501 or US-licensed adalimumab at a dose of 40 mg subcutaneously every other week. At week 24, there were no clinically meaningful differences in the safety, purity, or efficacy (by ACR20 responses) of the two drugs.
Study 263 was a randomized trial of ABP-501 in patients with plaque psoriasis and was conducted outside the United States. It included 350 patients with plaque psoriasis who received either 80 mg of EU-approved adalimumab or ABP 501 subcutaneously on day 1, then 40 mg every 2 weeks thereafter. The study's primary endpoint was the percent improvement in Psoriasis Area Severity Index (PASI) at week 16.
At week 16, patients in the EU-approved adalimumab group were randomly transitioned to ABP 501 or continued on EU-approved adalimumab through week 48. The transition from EU-approved adalimumab to ABP 501 showed no difference in the safety or immunogenicity profile of ABP 501.
The doses of ABP 501 submitted for approval will be similar to the originator Humira and will come as a single-use prefilled syringe and a single-use autoinjector at strengths (i.e., 20 mg/0.4 mL and 40 mg/0.8mL for the prefilled syringe, and 40 mg/0.8 mL for the autoinjector).
Safety and immunogenicity were also comparable, and the submission also included data to support the transition of adalimumab patients to the biosimilar, the firm previously noted.
Although FDA officials considered ABP-501 to be “highly similar” and the AAC panel discussion had concerns over extrapolation (especially for IBD where the mechanistic effects may not be similar), the overall vote was unanimous, 26-0, in favor of FDA approval of ABP-501. Interchangeability is not part of this application.
Issues
At issue is the potential introduction of multiple adalimumab biosimilars into the US market when Humiras' patent is slated to expire (December 2016).
There are numerous legal wranglings, by AbbVie and those making adalimumab biosimilars, challenging the Humira patent position. Abbvie’s multiple indications over the last few years may extend the patent as far as 2022. It is thought that the introduction of ADA biosimilars will have a substantial effect on Humiras current market position.
Continue Reading
ADD THE FIRST COMMENT
Disclosures
The author has received research/grant financial support on this subject
The author has received compensation as an advisor or consultant on this subject




If you are a health practitioner, you may Login/Register to comment.
Due to the nature of these comment forums, only health practitioners are allowed to comment at this time.