

2015 Guidelines for Idiopathic Pulmonary Fibrosis Save

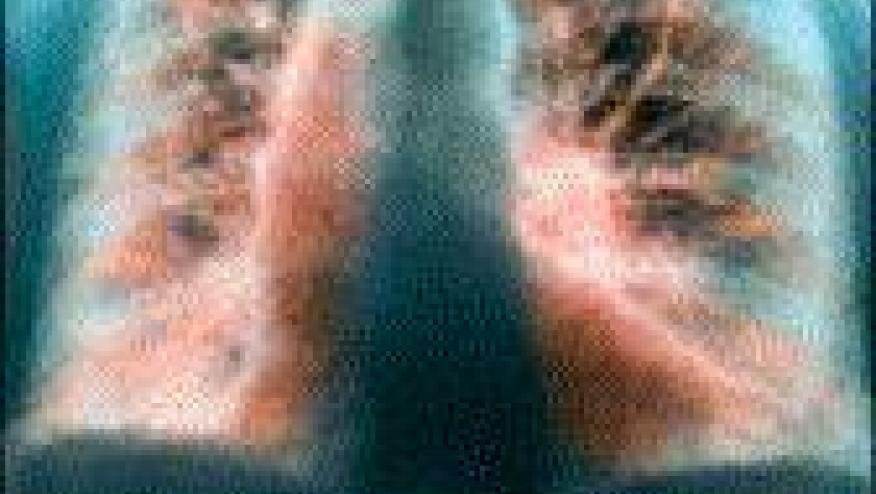
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrosing form of interstitial pneumonia, with poor survival rates of nearly 50% at 3 years. There are new joint guidelines for IPF from a conglomerate of international experts including the American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and the Latin American Thoracic Association (Citation source http://buff.ly/1D6V4A2).
The guidelines make strong or conditional recommendations for or against specific agents. Strong recommendations are made against the use of warfarin; imatinib (a selective tyrosine kinase inhibitor); and ambrisentan (a selective endothelin receptor antagonist). Conditional recommendations are made against sildenafil (a phosphodiesterase-5 inhibitor); and the dual endothelin receptor antagonists, macitentan and bosentan. Two new agents have been given conditional recommendations: nintedanib, a tyrosine kinase inhibitor; and pirfenidone, an oral antifibrotic drug. Pooled data on nintedanib show no significant effect on mortality or acute exacerbations of IPF, but a reduction in decline of forced vital capacity (FVC). Available trial data for pirfenidone show both a reduction in mortality and a reduced rate of FVC decline.
Continue Reading
ADD THE FIRST COMMENT
Disclosures
Disclosures
The author has no conflicts of interest to disclose related to this subject





If you are a health practitioner, you may Login/Register to comment.
Due to the nature of these comment forums, only health practitioners are allowed to comment at this time.