

Review of Amyloidosis Save
The New England Journal of Medicine has published a review of systemic amyloidosis.
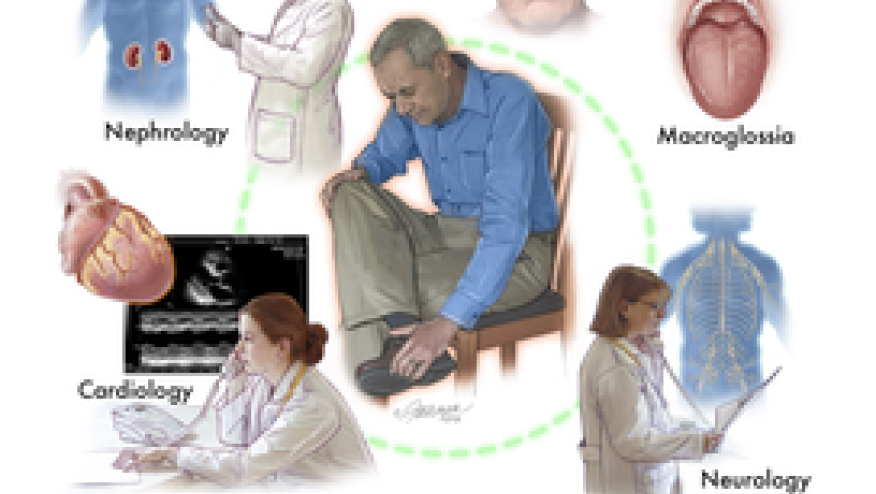
"Amyloidosis, a systemic disease that manifests in various ways, should be in the differential diagnosis of unexplained proteinuria, restrictive cardiomyopathy, peripheral and autonomic neuropathy, and hepatomegaly."
AL (immunoglobulin light chain) amyloidosis is a rare disease that often results in progressive organ dysfunction, organ failure and eventual death.
Clonal plasma cells in the bone marrow secrete free light chains into circulation. These light chains are part of immunoglobulins, also called antibodies. But in this disease, light chains misfold and aggregate into amyloid fibrils that deposit in organs and tissues.
In a review article of AL amyloidosis “Systemic Light Chain Amyloidosis,” Vaishali Sanchorawala, MD, director of the Amyloidosis Center at the Chobanian & Avedisian School of Medicine and Boston Medical Center, focused on recent advances in the understanding of pathogenesis, clinical syndromes, risk stratification and therapeutic advances, and looking at future efforts and needs in treatment and research.
“The care of patients with systemic immunoglobulin light chain (AL) amyloidosis has undergone transformative changes, leading to marked, steady progress in outcomes for patients over the past four decades,” Sanchorawala wrote in her review. “Overall survival has improved considerably, yet many unmet needs persist.”
“One of the most important determinants of survival is the severity of cardiac involvement,” said Sanchorawala. The heart is affected in 70 to 80% of patients with AL amyloidosis and cardiac problems are the leading cause of death, but other major organs like kidneys, liver, and peripheral and autonomic nervous systems can also be affected by this disease.
Therapies include those targeting clonal plasma cells, stopping light chain production, and new research into antifibril monoclonal antibodies that accelerate the removal of amyloid deposits from the organs.
“The arsenal of therapeutics for AL amyloidosis is rapidly expanding, offering a promising outlook and emerging triumph over adversity for patients in 2024,” said Sanchorawala. Early diagnosis is critical, and she recommended including evaluation for AL amyloidosis for patients exhibiting a wide range of symptoms and clinical syndromes.
“Despite improvements in the diagnosis and treatment of AL amyloidosis, continued basic and clinical research efforts are needed to brighten the future for patients with this disorder,” said Sanchorawala.
ADD THE FIRST COMMENT
Disclosures
Disclosures
The author has no conflicts of interest to disclose related to this subject






If you are a health practitioner, you may Login/Register to comment.
Due to the nature of these comment forums, only health practitioners are allowed to comment at this time.